Clustering and feature selection for single cell genomic data
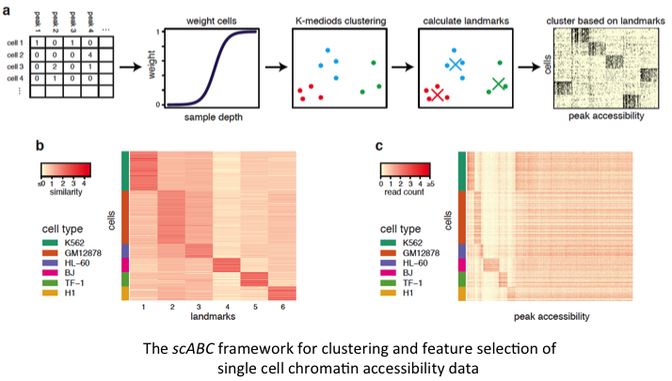
Recent advances in single cell technologies such as scATAC-seq and scChIP-seq have expanded our understanding of epigenetic heterogeneity at the single cell level. However, datasets arising from such technologies are difficult to analyze due to the inherent sparsity. In particular, consider scATAC-seq, designed to interrogate open chromatin in single cells. Open sites in a diploid genome have at most 2 chances to be captured through the assay and only a few thousand distinct reads are generated per cells, resulting in a very low chance that a particular site is captured by the assay. This creates a challenging task in delineating distinct sub-populations, as only a few genomic regions will have overlapping reads in a large number of cells.
To combat these challenges and allow for the de novo classification of individual cells by their epigenetic signatures, we present a statistical method for the unsupervised clustering of scATAC-seq data, named single cell Accessibility Based Clustering (scABC). In contrast to previous works that demand predefined accessible chromatin sites, our procedure relies solely on the patterns of read counts within genomic regions to cluster cells. It requires two inputs: the individual single cell mapped read files and the full set of called peaks (which can be obtained from the union of all of the individual cells without the need for additional experiments). We apply our method to publicly available scATAC-seq data, as well as a true biological mixture to show that our approach can cluster cells with similar epigenetic patterns and identify accessible regions specific to each cluster. We further demonstrate that the cluster specific accessible regions determined by scABC have functional meaning and are capable of determining cellular identity. In particular, we show that these cluster specific accessible regions are enriched for transcription factor motifs known to be specific to each subpopulation and that, through association with scRNA-seq data, they can lead to the identification of subpopulation specific gene expression.
Publication
To combat these challenges and allow for the de novo classification of individual cells by their epigenetic signatures, we present a statistical method for the unsupervised clustering of scATAC-seq data, named single cell Accessibility Based Clustering (scABC). In contrast to previous works that demand predefined accessible chromatin sites, our procedure relies solely on the patterns of read counts within genomic regions to cluster cells. It requires two inputs: the individual single cell mapped read files and the full set of called peaks (which can be obtained from the union of all of the individual cells without the need for additional experiments). We apply our method to publicly available scATAC-seq data, as well as a true biological mixture to show that our approach can cluster cells with similar epigenetic patterns and identify accessible regions specific to each cluster. We further demonstrate that the cluster specific accessible regions determined by scABC have functional meaning and are capable of determining cellular identity. In particular, we show that these cluster specific accessible regions are enriched for transcription factor motifs known to be specific to each subpopulation and that, through association with scRNA-seq data, they can lead to the identification of subpopulation specific gene expression.
Publication
- Zamanighomi M*, Lin Z*, Daley T*, Chen Xi , Zhana Duren, Schep A, Greenleaf WJ, and Wong WH: Unsupervised clustering and epigenetic classification of single cells. Nature Communications. 2018 (In print). [paper link] [software link]